


주메뉴
- About IBS 연구원소개
-
Research Centers
연구단소개
- Research Outcomes
- Mathematics
- Physics
- Center for Underground Physics
- Center for Theoretical Physics of the Universe (Particle Theory and Cosmology Group)
- Center for Theoretical Physics of the Universe (Cosmology, Gravity and Astroparticle Physics Group)
- Dark Matter Axion Group
- Center for Artificial Low Dimensional Electronic Systems
- Center for Theoretical Physics of Complex Systems
- Center for Quantum Nanoscience
- Center for Exotic Nuclear Studies
- Center for Van der Waals Quantum Solids
- Center for Relativistic Laser Science
- Chemistry
- Life Sciences
- Earth Science
- Interdisciplinary
- Center for Neuroscience Imaging Research (Neuro Technology Group)
- Center for Neuroscience Imaging Research (Cognitive and Computational Neuroscience Group)
- Center for Algorithmic and Robotized Synthesis
- Center for Genome Engineering
- Center for Nanomedicine
- Center for Biomolecular and Cellular Structure
- Center for 2D Quantum Heterostructures
- Center for Quantum Conversion Research
- Institutes
- Korea Virus Research Institute
- News Center 뉴스 센터
- Career 인재초빙
- Living in Korea IBS School-UST
- IBS School 윤리경영

주메뉴
- About IBS
-
Research Centers
- Research Outcomes
- Mathematics
- Physics
- Center for Underground Physics
- Center for Theoretical Physics of the Universe (Particle Theory and Cosmology Group)
- Center for Theoretical Physics of the Universe (Cosmology, Gravity and Astroparticle Physics Group)
- Dark Matter Axion Group
- Center for Artificial Low Dimensional Electronic Systems
- Center for Theoretical Physics of Complex Systems
- Center for Quantum Nanoscience
- Center for Exotic Nuclear Studies
- Center for Van der Waals Quantum Solids
- Center for Relativistic Laser Science
- Chemistry
- Life Sciences
- Earth Science
- Interdisciplinary
- Center for Neuroscience Imaging Research (Neuro Technology Group)
- Center for Neuroscience Imaging Research (Cognitive and Computational Neuroscience Group)
- Center for Algorithmic and Robotized Synthesis
- Center for Genome Engineering
- Center for Nanomedicine
- Center for Biomolecular and Cellular Structure
- Center for 2D Quantum Heterostructures
- Center for Quantum Conversion Research
- Institutes
- Korea Virus Research Institute
- News Center
- Career
- Living in Korea
- IBS School





News Center

Charting a path toward overcoming glioblastoma resistance to chemotherapy- Mutational signatures etched into the cells' genome by an anti-cancer drug called temozolomide (TMZ) uncover an Achilles’ heel for TMZ chemotherapy resistance - In spite of intensive research, glioblastoma remains one of the most lethal types of brain cancer. Temozolomide (TMZ) is used as the front-line medicine in its treatment. While TMZ effectively penetrates the brain and targets tumors, its success depends on the tumor cells attempting to repair the DNA damage caused by the drug. Unfortunately, glioblastomas often evade treatment by inactivating the various DNA repair pathways, making them resistant to TMZ and limiting its effectiveness. In these drug-resistant cancer cells, DNA becomes mutated but does not lead to cell death. The researchers at the Center for Genomic Integrity within the Institute for Basic Science (IBS) in Ulsan, South Korea, in conjunction with the bioinformatics team from the Ulsan National Institute of Science and Technology (UNIST), have uncovered critical insights into the mechanisms behind TMZ resistance. Their work could pave the way for more effective therapies against this devastating cancer. Understanding How TMZ Works—and Fails TMZ works by introducing DNA damage, specifically a modification called O6-methyl guanine (O6-meG), which is a modified DNA base guanine with a methyl group added to the oxygen in position 6. Normally, a cell's mismatch repair (MMR) system attempts to fix the damage, but in the case of O6-meG, the mutated base pair can efficiently pair with thymine as with cytosine. This causes the repair process to go awry, creating a vicious cycle of failed repair attempts that ultimately kill the tumor cells. However, if the MMR pathway is disabled, O6-meG no longer triggers this toxic cycle. Instead, it leads to a massive number of cytosine-to-thymine mutations without killing the cells. The tumor with defective MMR becomes 100 times more resistant to TMZ. It is possible to still kill these resistant tumors by administering a very high dose of TMZ. At these high concentrations, TMZ generates another methylated base, 3-methyl adenine (3-meA), which blocks the DNA synthesis in cancer cells. This base is repaired by a different DNA repair pathway, called base excision repair (BER). The first enzyme in the BER pathway, called MPG, excises not the entire nucleotide but only the base part of it, creating an abasic site, which is converted into a single-strand DNA break by another enzyme called APE1, and the gap is then filled and sealed. However, if APE1 is inhibited, glioblastoma cells become highly sensitive to TMZ, even if the MMR pathway is inactive. Thus, APE1 represents the Achilles’ heel (i.e., the most vulnerable spot) of tumor chemoresistance. Surprising Insight into Mutations and its Relation to Aging Surprisingly, the IBS researchers found that if the MPG enzyme is inactivated and BER cannot be initiated, the cells remain resistant to TMZ. This is because the replication block can be overcome with the help of a specialized polymerase that can insert adenine in place of the blocking DNA residue. Using the whole genome sequencing, the IBS/UNIST team could detect a mutational “scar” marking the spot where the replication block occurred. The specialized DNA polymerases that come to the rescue when the DNA replication is blocked by 3-meA or any other replication-blocking lesion are aptly called translesion synthesis (TLS) polymerases. They differ from the main replicative enzymes, which synthesize the bulk of DNA, in that they are less precise and can insert mismatched nucleotides, which allows them to bypass the lesion. However, this unique feature can have undesired consequences: TLS polymerases not only bypass the roadblocks on the way of replication, they also introduce errors. The more often the cell has to employ TLS polymerases, the more mutational “scars” accumulate in the genome. One particular TLS polymerase, so-called polymerase zeta, is called to the rescue of the stalled replication forks more often than others. It has its own “mutational signature”, that is etched into the genome wherever polymerase zeta is active. IBS researchers discovered that polymerase zeta increases mutational background in TMZ-treated cells. Importantly, apart from contributing to the mutational burden following the TMZ treatment, polymerase zeta was found in this study to also be the main culprit responsible for the mutation accumulation in untreated cells. As organisms age, their cells accumulate mutations. There is a striking correlation between the rate of mutation accumulation and the organism’s lifespan, e.g., a short-living mouse accumulates mutations faster than a long-living human. The mutation patterns left behind by polymerase zeta happen to resemble one of the mutational patterns found in aging mammals. This unexpected finding sheds light on one of the potential mechanisms of aging. Charting a Path Forward The IBS researchers used a comprehensive collection of DNA repair mutants to study which genes are required to survive the TMZ treatment. They analyzed TMZ sensitivities of dozens of cell lines, each having one DNA repair gene inactivated. They also sequenced more than 400 genomes of treated and untreated cells to determine what mutations were caused by the DNA repair pathway inactivation, by the TMZ treatment, and by their combination. Bioinformatics analysis of mutations reveals so-called “mutational signatures”, which can be caused by the chemical substances, irradiation, and inactivation of DNA repair genes, which frequently occurs in cancer. Computer programs analyze all the mutations found in the genome and extract mutational patterns primarily based on the nucleotides to the left and to the right of the nucleotide substitutions. This study is the first to use a comprehensive approach using a collection of knockouts in the normal and MMR deficient genetic backgrounds and combining cell survival assays with whole genome sequencing. In the course of this research, it became clear that DNA repair pathways underlying drug resistance are redundant, i.e., when one pathway is inactivated, another can serve as a backup. To reveal this redundancy, it is necessary to generate multiple knockouts, sequentially inactivating different pathways, not unlike peeling an onion. As the cell is stripped of its DNA repair defenses, layer by layer, the genomic mutational signatures change, reflecting which mechanisms are activated in response to the drug (Fig. 1). One of the findings is that the inactivation of some genes, e.g., FANCD2, further sensitizes the MMR-competent cells to TMZ but doesn’t affect the resistance of MMR-deficient cells. On the contrary, knockout of genes involved in BER, such as APE1 and XRCC1, sensitizes MMR-deficient cells but has very little effect on MMR-proficient ones (Fig.2). The study makes it possible to outline the path to combat TMZ resistance with the DNA repair protein inhibitors. For example, substances inhibiting MPG by themselves are not likely to make the TMZ effect more potent. On another hand, inhibition of APE1 appears to be a very promising approach to combat the build-up of TMZ resistance. As the drugs targeting APE1 are being developed, it will be important to test them for synergistic effects with TMZ. Another potentially promising approach is a combination of APE1 and TLS inhibitors. The IBS/UNIST researchers now plan to focus on identifying TLS polymerases that are relevant for TMZ resistance (Fig.3). The findings of the IBS/UNIST team represent a significant step forward in understanding glioblastoma resistance and offer hope for new, more effective therapies. As the researchers continue to explore the intricate layers of tumor defenses, their work brings us closer to developing treatments that can outsmart even the most stubborn cancers.
Notes for editors
- References
- Media Contact
- About the Institute for Basic Science (IBS)
|
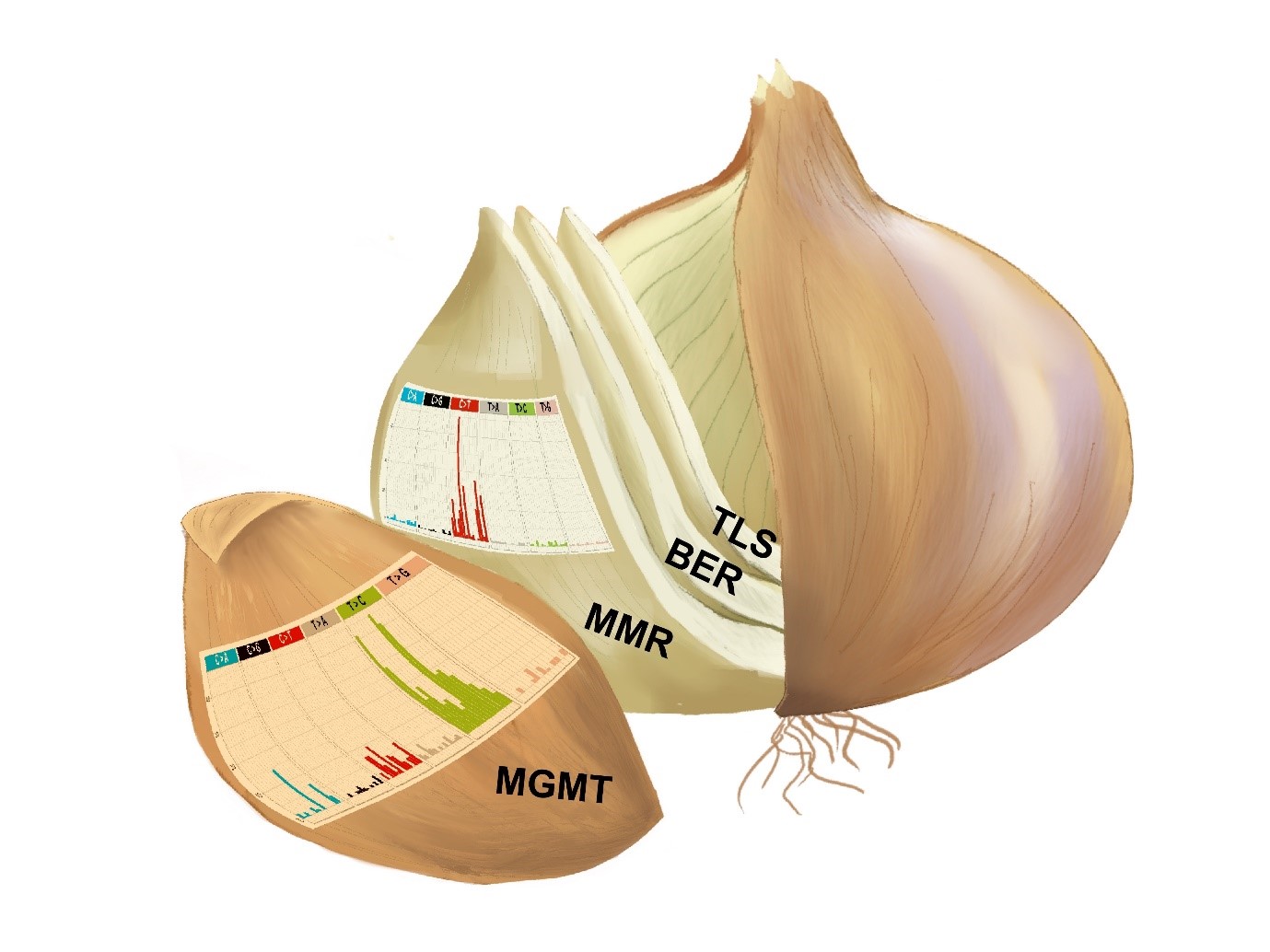 Figure 1. The “onion model” illustrates how the one-by-one inactivation of the DNA repair pathways brings about changes in TMZ-induced mutational signatures. Every “leaf” of the onion represents the cell line and the graph illustrates the classes of single nucleotide substitutions caused by TMZ. The outer layer is a normal human cell and the second layer is the TMZ-sensitive tumor. Note how different are the TMZ mutational signatures in these two cell types.
Figure 1. The “onion model” illustrates how the one-by-one inactivation of the DNA repair pathways brings about changes in TMZ-induced mutational signatures. Every “leaf” of the onion represents the cell line and the graph illustrates the classes of single nucleotide substitutions caused by TMZ. The outer layer is a normal human cell and the second layer is the TMZ-sensitive tumor. Note how different are the TMZ mutational signatures in these two cell types.
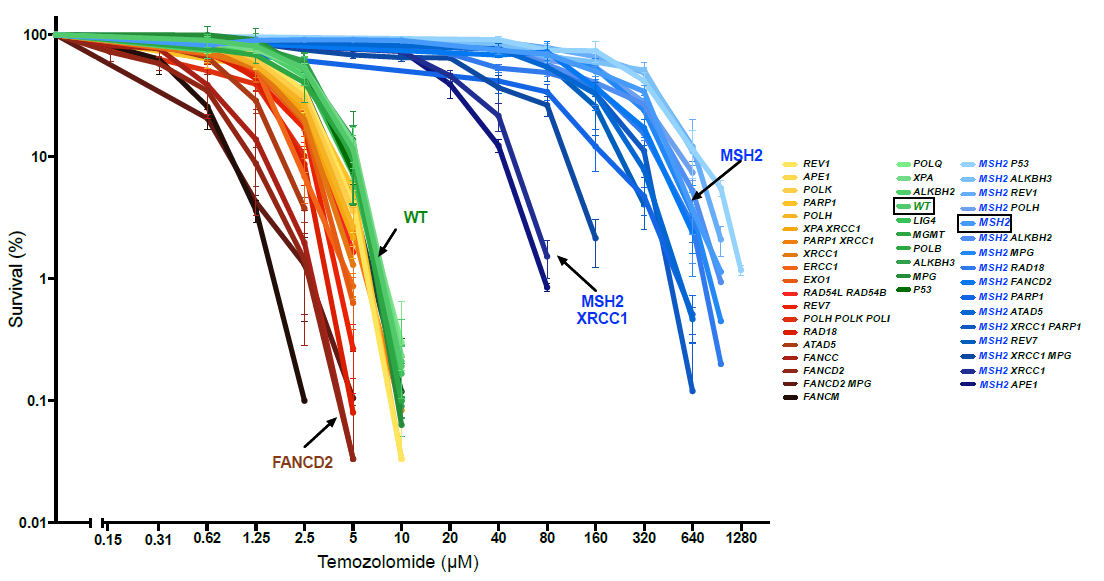 Figure 2. The survival curves of different DNA repair mutants treated with increasing TMZ concentrations. In this graph, the WT (green) is the TMZ-sensitive tumor. The blue lines are the MMR-deficient mutants, which are a hundred times more resistant than the WT line. Note how knockout of XRCC1 and APE1 genes restores the TMZ sensitivity of the MMR mutants.
Figure 2. The survival curves of different DNA repair mutants treated with increasing TMZ concentrations. In this graph, the WT (green) is the TMZ-sensitive tumor. The blue lines are the MMR-deficient mutants, which are a hundred times more resistant than the WT line. Note how knockout of XRCC1 and APE1 genes restores the TMZ sensitivity of the MMR mutants.
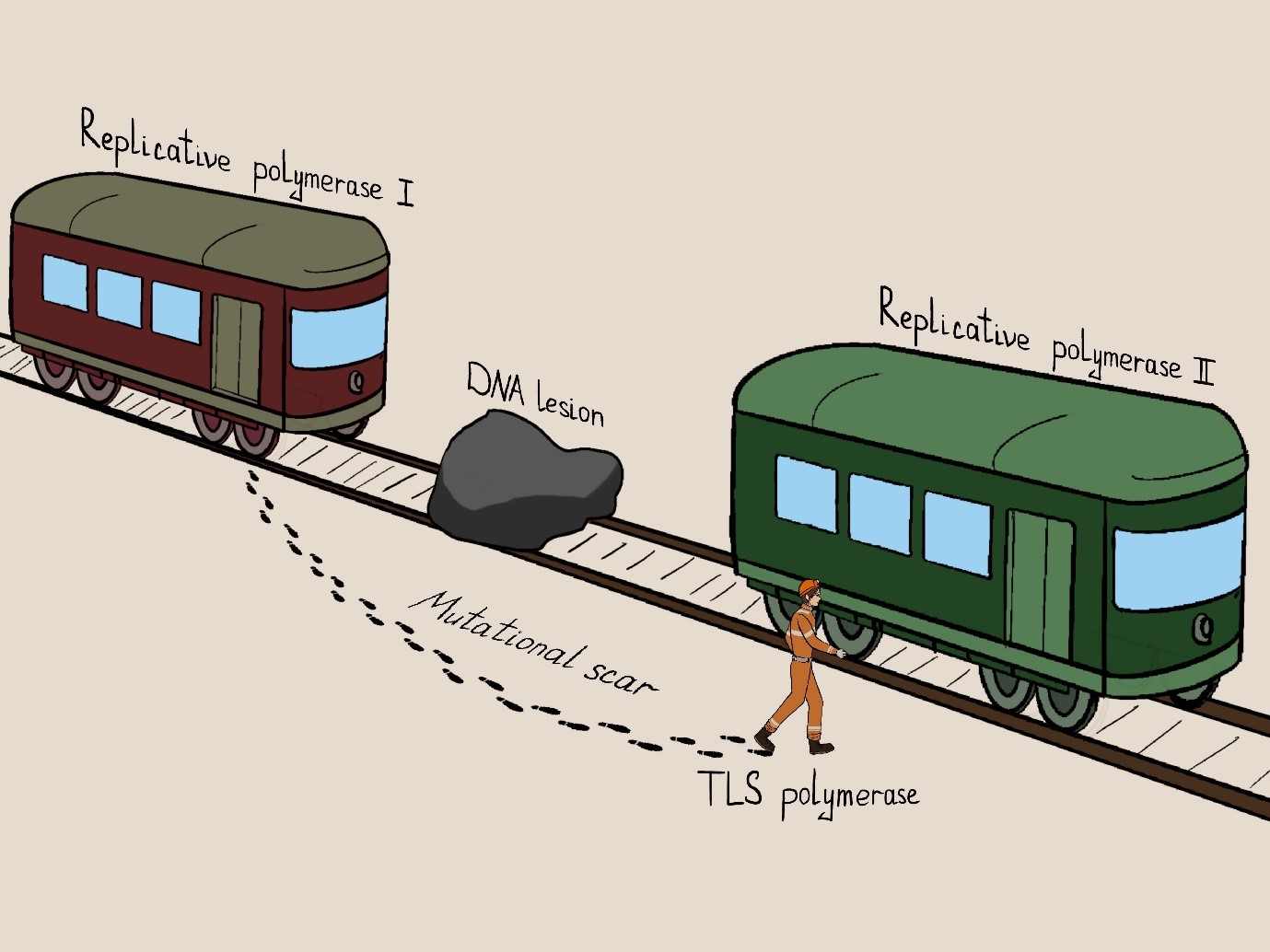 Figure 3. An artistic representation of the mutational “scar” left by translesion synthesis. When a train (replicative polymerase) encounters an obstacle, the passenger has to disembark and proceed around the obstacle on foot (error-prone translesion polymerase), leaving footprints (mutations). These mutations are detected by whole genome sequencing and mark the locations where the switch to a translesion polymerase occurred.
Figure 3. An artistic representation of the mutational “scar” left by translesion synthesis. When a train (replicative polymerase) encounters an obstacle, the passenger has to disembark and proceed around the obstacle on foot (error-prone translesion polymerase), leaving footprints (mutations). These mutations are detected by whole genome sequencing and mark the locations where the switch to a translesion polymerase occurred.
| Next | |
|---|---|
| before |
- Content Manager
- Public Relations Team : Yim Ji Yeob 042-878-8173
- Last Update 2023-11-28 14:20

55, Expo-ro, Yuseong-gu, Daejeon, Korea, 34126
Tel. +82-42-878-8114 | Fax. +82-42-878-8079 | E-mail. webmaster@ibs.re.kr
Copyright(c) IBS. All Rights Reserved.
