


주메뉴
- About IBS 연구원소개
-
Research Centers
연구단소개
- Research Outcomes
- Mathematics
- Physics
- Center for Underground Physics
- Center for Theoretical Physics of the Universe (Particle Theory and Cosmology Group)
- Center for Theoretical Physics of the Universe (Cosmology, Gravity and Astroparticle Physics Group)
- Dark Matter Axion Group
- Center for Artificial Low Dimensional Electronic Systems
- Center for Theoretical Physics of Complex Systems
- Center for Quantum Nanoscience
- Center for Exotic Nuclear Studies
- Center for Van der Waals Quantum Solids
- Center for Relativistic Laser Science
- Chemistry
- Life Sciences
- Earth Science
- Interdisciplinary
- Center for Neuroscience Imaging Research (Neuro Technology Group)
- Center for Neuroscience Imaging Research (Cognitive and Computational Neuroscience Group)
- Center for Algorithmic and Robotized Synthesis
- Center for Genome Engineering
- Center for Nanomedicine
- Center for Biomolecular and Cellular Structure
- Center for 2D Quantum Heterostructures
- Center for Quantum Conversion Research
- Institutes
- Korea Virus Research Institute
- News Center 뉴스 센터
- Career 인재초빙
- Living in Korea IBS School-UST
- IBS School 윤리경영

주메뉴
- About IBS
-
Research Centers
- Research Outcomes
- Mathematics
- Physics
- Center for Underground Physics
- Center for Theoretical Physics of the Universe (Particle Theory and Cosmology Group)
- Center for Theoretical Physics of the Universe (Cosmology, Gravity and Astroparticle Physics Group)
- Dark Matter Axion Group
- Center for Artificial Low Dimensional Electronic Systems
- Center for Theoretical Physics of Complex Systems
- Center for Quantum Nanoscience
- Center for Exotic Nuclear Studies
- Center for Van der Waals Quantum Solids
- Center for Relativistic Laser Science
- Chemistry
- Life Sciences
- Earth Science
- Interdisciplinary
- Center for Neuroscience Imaging Research (Neuro Technology Group)
- Center for Neuroscience Imaging Research (Cognitive and Computational Neuroscience Group)
- Center for Algorithmic and Robotized Synthesis
- Center for Genome Engineering
- Center for Nanomedicine
- Center for Biomolecular and Cellular Structure
- Center for 2D Quantum Heterostructures
- Center for Quantum Conversion Research
- Institutes
- Korea Virus Research Institute
- News Center
- Career
- Living in Korea
- IBS School





News Center

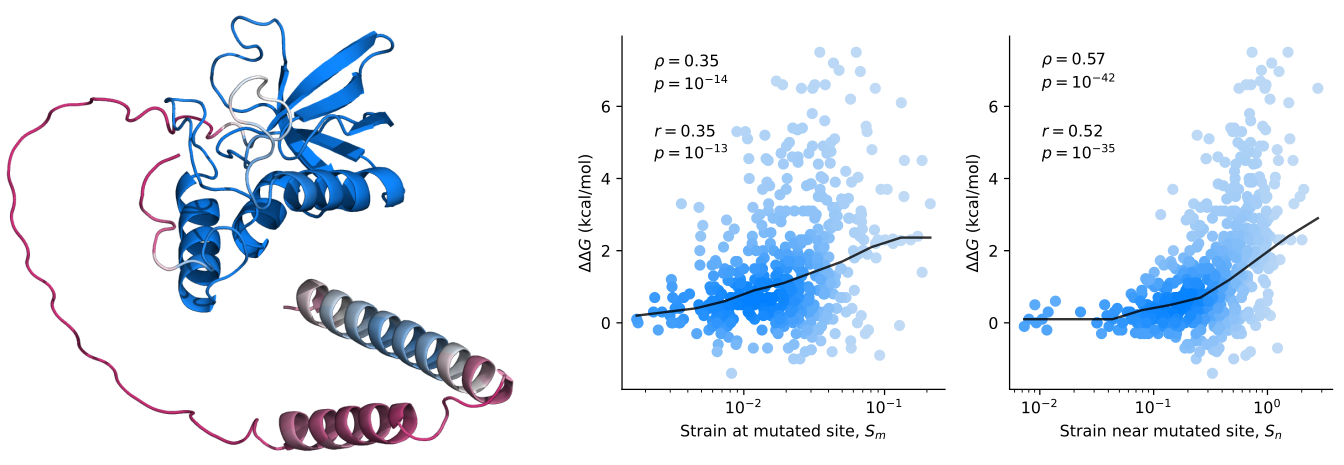
Protein mutant stability can be inferred from AI-predicted structures- AlphaFold2 predicts structural effects of mutations that are correlated with changes in stability - Researchers at the Center for Algorithmic and Robotized Synthesis within the Institute for Basic Science have taken a significant step forward in understanding the stability of proteins by leveraging the power of AI. The research team used AlphaFold2 to explore how mutations affect protein stability—a crucial factor in ensuring proteins function correctly and do not cause diseases like Alzheimer's. DeepMind’s AlphaFold algorithm, which can accurately predict a protein’s structure from its gene, has been a game-changer across the field of biology, making structural biology accessible to everyone. Despite this immense success, two fundamental questions remain unanswered: Will the predicted structures fold correctly and stay folded? And a general question about AI algorithms: how does AlphaFold actually work? A critical limitation of AlphaFold is that it was trained on a set of stable proteins that stay folded at physiological temperatures. As a result, it predicts the most likely folded structure without knowing if it will certainly fold or will be unstable. Knowing and predicting protein stability is crucial because unstable proteins can misfold, leading to dysfunction and potentially serious diseases, so the cells must spend much energy to get rid of them. Furthermore, most proteins are only marginally stable, making them highly susceptible to mutations that can cause them to unfold. Thus, protein engineering is much about careful navigation in a minefield of dysfunctional protein sequences that do not fold. All this implies that the next step in using AlphaFold should be to try to predict those changes in stability due to mutations. A fundamental question tested in this study was whether AlphaFold has learned the underlying physics of protein folding or is simply a high-dimensional regression machine that merely recognizes statistical patterns. This question is about the capacity to generalize: if AlphaFold has somehow learned the physical forces in action, it ought to work on protein sequences it has not seen before. That’s exactly what the two IBS researchers, John MCBRIDE and Tsvi TLUSTY, wanted to test in their study. Their way to tackle this question was to examine if AlphaFold can correctly predict the effects of mutations on stability. There are infinitely more mutations than data points used in the training of AlphaFold, meaning that even very sophisticated regression will not suffice to account for the full range of mutation effects. This task is highly challenging since critical changes in stability often involve small structural changes that are hard to predict. Still, it turns out that there are some useful clues within the structural changes predicted by AlphaFold that provide valuable information on possible stability changes. IBS researchers showed this by comparing structural changes inflicted by mutations to differences in experimentally measured stability between the wild-type and mutated protein [1]. A critical ingredient was using a probe that is very sensitive to small changes. The researchers devised an innovative metric, known as the effective strain, [2] to detect small but important changes in protein structure that are linked to stability. Looking at thousands of mutations, they found that the effective strain measure correlates with the magnitude of the change in stability. That is to say, large structural changes (predicted by AlphaFold) also predict large changes in stability. Lead author John MCBRIDE stated, “This is a strong indication that the structures predicted by AlphaFold encode significant physical information, particularly about stability. It is necessary to develop new physical models to decode this information further.” These insights open up new possibilities for protein engineering, a field that involves designing proteins with specific functions. By better understanding how mutations affect stability, scientists can navigate the complex landscape of protein design more effectively, potentially leading to advances in drug development and treatments for diseases caused by protein misfolding. This research marks an important milestone in the ongoing exploration of how AI can be used to unravel the complexities of biology and underscores the need for further studies to fully unlock the potential of AI in scientific discovery.
Notes for editors
- References
- Media Contact
- About the Institute for Basic Science (IBS)
|
| Next | |
|---|---|
| before |
- Content Manager
- Public Relations Team : Yim Ji Yeob 042-878-8173
- Last Update 2023-11-28 14:20

55, Expo-ro, Yuseong-gu, Daejeon, Korea, 34126
Tel. +82-42-878-8114 | Fax. +82-42-878-8079 | E-mail. webmaster@ibs.re.kr
Copyright(c) IBS. All Rights Reserved.
